


ממומן
Understanding SMA: The Genetic Disease Changing with Gene-Based Therapies
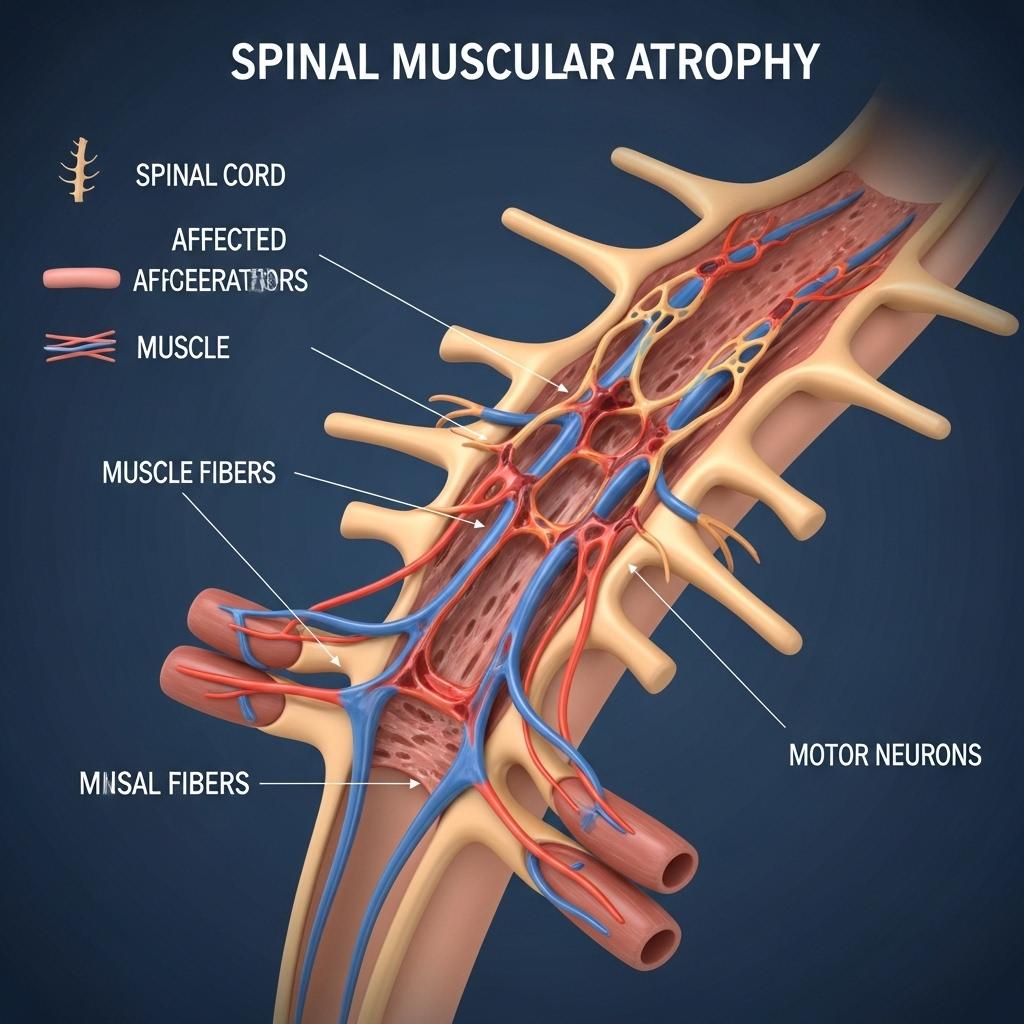
Spinal Muscular Atrophy (SMA) is a severe, genetically inherited neuromuscular disorder characterized by progressive muscle atrophy and weakness due to degeneration of alpha motor neurons in the spinal cord. Once considered a leading genetic cause of infant mortality, SMA has undergone a radical transformation in recent years, driven by advances in genetic science and the development of disease-modifying therapies.
Understanding SMA at the molecular level has enabled targeted treatment approaches, dramatically altering outcomes for affected individuals and bringing renewed hope to families and clinicians.
Request a sample copy of the CI report at: https://www.datamintelligence.com/strategic-insights/ci/spinal-muscular-atrophy-genetic-understanding-disease-modifying-therapies
Genetic Basis and Inheritance
SMA is primarily caused by biallelic mutations (deletions or variants) in the SMN1 gene (Survival Motor Neuron 1) located on chromosome 5q13. The absence or dysfunction of the SMN1 gene product leads to degeneration of motor neurons, ultimately impairing voluntary muscle function.
Humans also possess a paralog gene SMN2, which can partially compensate for the loss of SMN1. However, SMN2 produces only a fraction of functional SMN protein due to alternative splicing that excludes exon 7. The number of SMN2 copies strongly influences disease severity: fewer copies correlate with more severe phenotypes.
SMA is inherited in an autosomal recessive manner, meaning both parents must carry a defective SMN1 gene for a child to be affected. Carrier frequency is approximately 1 in 40 to 1 in 60, making population screening increasingly relevant.
SMA Classification and Disease Severity
SMA is classified based on age of onset and motor milestones achieved:
* Type 0: Prenatal onset, respiratory failure at birth (rare, severe)
* Type 1 (Werdnig-Hoffmann): Onset <6 months; never sit independently; high early mortality without treatment
* Type 2: Onset between 6–18 months; sit but never walk unaided
* Type 3 (Kugelberg-Welander): Childhood/adolescence onset; can walk but experience progressive weakness
* Type 4: *** onset with mild symptoms and slow progression
Type 1 is the most common and severe form, accounting for approximately 50–60% of cases.
Diagnostic Evolution: From Clinical Suspicion to Genetic Confirmation
While clinical features such as hypotonia, delayed milestones, and muscle weakness raise suspicion, genetic testing confirms the diagnosis by detecting SMN1 gene deletions and assessing SMN2 copy number. Electromyography (EMG) and muscle biopsy are rarely needed in the molecular era.
Newborn screening (NBS) programs have now been implemented in many regions, allowing pre-symptomatic diagnosis and early intervention, critical for maximizing therapeutic efficacy.
Breakthrough Therapies: From Supportive to Disease-Modifying
The therapeutic landscape for SMA has shifted dramatically in less than a decade.
Three FDA-approved disease-modifying therapies are now available:
* Nusinersen (Spinraza): An antisense oligonucleotide administered intrathecally; it modifies SMN2 splicing to increase full-length SMN protein.
* Onasemnogene abeparvovec-xioi (Zolgensma): A one-time AAV9-based gene therapy delivering a functional copy of SMN1; approved for patients under age 2.
* Risdiplam (Evrysdi): An oral SMN2 splicing modifier approved for patients ≥2 months; enables systemic increase in SMN protein.
These therapies have significantly improved survival, motor function, and quality of life, especially when administered early. However, long-term safety, durability, and access remain areas of continued observation and discussion.
Remaining Challenges and Emerging Research
Despite treatment gains, SMA still poses unmet clinical and health policy challenges:
* Delayed diagnosis in late-onset types due to milder symptoms
* High costs of therapies, prompting global health access concerns
* Variable response to treatment due to age, disease severity, and SMN2 copy number
Research efforts are focused on:
* Combination therapies (e.g., gene therapy + oral agents)
* Neuroprotective strategies to preserve motor function
* Muscle-targeted approaches such as myostatin inhibitors
* Biomarker development for treatment monitoring
Request a CI consultation at: https://www.datamintelligence.com/strategic-insights/ci/spinal-muscular-atrophy-genetic-understanding-disease-modifying-therapies
Genetic Counseling and Family Planning
Carrier screening is crucial in families with a SMA history or populations with high carrier frequency. Genetic counseling helps at-risk couples make informed reproductive choices, including options like prenatal testing or preimplantation genetic diagnosis (PGD).
As SMA management transitions into a chronic care model, integrating genetic support with neurologic care remains essential for optimizing lifelong outcomes.
About DataM Intelligence
DataM Intelligence 4Market Research LLP delivers real-time competitive intelligence across autoimmune, immunologic, and rare disease spaces. Our insights span clinical pipelines, regulatory benchmarks, and commercialization strategies for stakeholders in global life sciences.
🔗 Visit: www.datamintelligence.com
ממומן
ממומן
ממומן
קטגוריות
Read More
If you’re a small business owner, reseller, custom t-shirt printer, or someone launching your own clothing brand, there’s one thing you’ve probably searched online more than once: bulk apparel Chicago. And you’re not alone. The demand for wholesale, bulk clothing in Chicago has grown tremendously in recent years. Whether you're looking to stock up for your shop, fulfill...

New Zenith replica watches Chronomaster Original Triple Calendar (Lapis Lazuli) A uneven natural stone dial brings together the technically advanced El Primero calendar edition. The Zenith Triple Calendar is based on the lightweight 38mm case of the Chronomaster Original, which itself is dependent on the 1969 El Uno A386 model. This ***gn provided a balanced and...

In the realm of automotive accessories, car wheel caps might seem like a minor detail, but they play a significant role in vehicle aesthetics and functionality. As the global automotive market evolves, so does the demand and innovation surrounding wheel caps. This article explores the latest trends and developments in the global car wheel cap market, highlighting key innovations, market...
🌐 CLICK HERE 🟢==►► WATCH NOW 🔴 CLICK HERE 🌐==►► Download Now https://***.my.id/watch-streaming/?video=watchat4k-chonera-bonita-video-video-de-la

無論您是電子煙新手還是經驗豐富的使用者,了解不同類型電子煙的壽命對於規劃使用成本至關重要。從一次性電子煙到可加油和可替換電池的設備,每種電子煙的壽命都取決於設計、電池容量和使用方式。本文將詳細介紹不同類型電子煙的使用壽命及其影響因素,並提供延長設備使用壽命的建議。 一次性電子煙的使用壽命 一次性電子煙的壽命主要取決於其內部的煙油容量和使用頻率。通常,一次性電子煙可使用幾天,直到煙油耗盡。 吸入次數(Puff Count):每款一次性電子煙包裝上都標註了預估吸入次數,然而,實際吸入次數可能因吸煙習慣而異,長時間吸入會減少可用次數。 煙油容量與尼古丁含量:煙油容量越大,尼古丁濃度越高,使用時間越長。 預填充煙彈電子煙的使用壽命 預填充煙彈電子煙由可更換的煙彈和可充電的設備組成。設備的壽命取決於電池性能,而煙彈通常可使用1至2天。...