

How Pharmacologic Therapies Are Transforming PKU Treatment
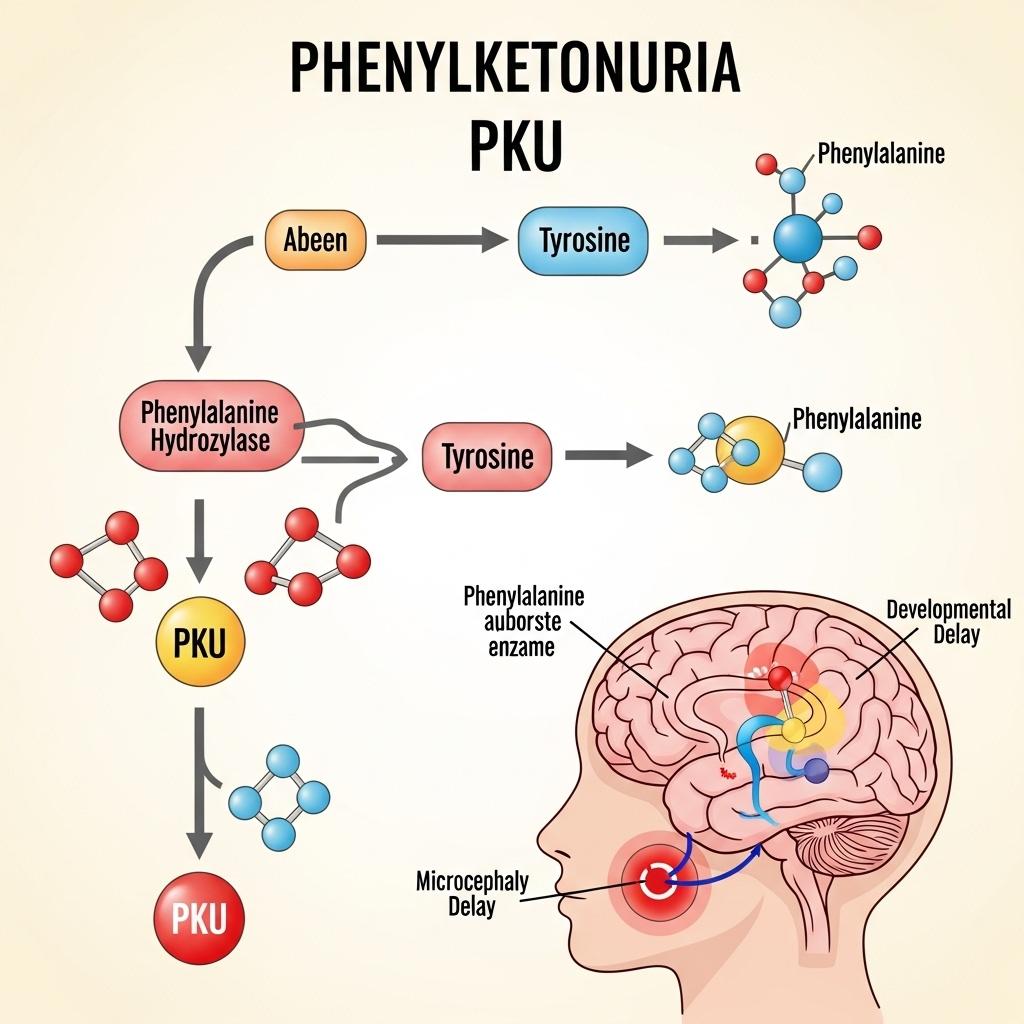
Phenylketonuria (PKU) is a rare autosomal recessive metabolic disorder caused by mutations in the PAH gene, leading to a deficiency in the enzyme phenylalanine hydroxylase. This enzyme is essential for converting phenylalanine (Phe)—an amino acid found in many protein-containing foods—into tyrosine. Without functional PAH, phenylalanine accumulates in the blood and brain, becoming neurotoxic.
Request a sample copy of the CI report at: https://www.datamintelligence.com/download-sample/phenylketonuria-treatment-market
Early identification and lifelong management of PKU are critical to preventing intellectual disability, seizures, behavioral issues, and other neurological impairments. Advances in newborn screening, medical nutrition therapy, and emerging pharmacologic treatments are redefining the PKU care pathway.
Genetic Basis and Global Epidemiology
PKU is one of the most well-characterized inborn errors of metabolism. It affects approximately 1 in 10,000 to 15,000 live births globally, with higher prevalence in certain regions such as Turkey, Ireland, and parts of the Middle East. The severity of PKU depends on residual PAH enzyme activity, with the classic form being the most severe.
Mutations in the PAH gene on chromosome 12 account for over 98% of PKU cases. Rarely, defects in the cofactor tetrahydrobiopterin (BH4) metabolism can produce similar symptoms and are termed "non-classic" or BH4-deficient PKU.
Newborn Screening and Early Diagnosis
Widespread adoption of newborn screening programs using tandem mass spectrometry enables early detection within the first few days of life. Elevated phenylalanine levels prompt confirmatory testing, followed by immediate dietary intervention to prevent neurocognitive damage.
Early-treated individuals often develop normally if therapy is maintained rigorously. However, neuropsychological challenges can still arise due to subclinical Phe fluctuations, especially during adolescence and adulthood.
Medical Nutrition Therapy: Foundation of PKU Management
The cornerstone of PKU treatment is a lifelong low-phenylalanine diet. This typically involves:
* Phe-restricted natural foods: avoiding high-protein items like meat, dairy, eggs, legumes, and nuts.
* Specialty low-protein foods: bread, pasta, and cereals formulated for PKU patients.
* Medical formula: amino acid mixtures devoid of Phe to provide adequate protein, vitamins, and minerals.
* Maintaining blood Phe levels within the 120–360 µmol/L range is critical, particularly for children and during pregnancy. Adherence is challenging, especially during adolescence and adulthood, contributing to poor metabolic control and neurocognitive risks.
Pharmacologic Therapies: Toward Less Dietary Restriction
Emerging therapies aim to reduce dietary burden and improve metabolic stability. Approved pharmacologic options include:
1. Sapropterin dihydrochloride (Kuvan®): A synthetic form of BH4 that increases residual PAH activity in responsive patients (~20–50% of cases). It enables some individuals to tolerate higher Phe intake.
2. Pegvaliase (Palynziq®): An injectable enzyme substitution therapy that breaks down Phe in the bloodstream. Approved for adults with uncontrolled blood Phe levels despite dietary management. Pegvaliase significantly reduces Phe levels and allows liberalized diets but requires monitoring for hypersensitivity.
Gene therapy and genome editing are being studied as potential curative approaches, with early-phase trials exploring AAV-mediated PAH gene delivery to the liver.
PKU in Women and Maternal Phenylketonuria
Women with PKU require strict Phe control before and during pregnancy, as elevated Phe levels can cause congenital heart defects, intellectual disability, and microcephaly in the fetus, even if the child does not inherit PKU.
Maternal PKU syndrome remains a critical area of focus, with metabolic clinics emphasizing preconception counseling and tight dietary control to prevent teratogenic effects.
Read the full CI Insights report: https://www.datamintelligence.com/strategic-insights/phenylketonuria-pku
Psychosocial Impact and Long-Term Care
Even with good metabolic control, individuals with PKU may experience:
* Executive function deficits
* Mood disorders such as anxiety or depression
* Reduced quality of life from strict dietary adherence
Lifelong follow-up, neuropsychological assessments, and support from multidisciplinary metabolic teams—including dietitians, psychologists, and social workers—are essential for optimizing outcomes.
Digital tools, telehealth platforms, and metabolic mobile apps are increasingly integrated into care models to support adherence, track Phe levels, and enhance patient engagement.
About DataM Intelligence
DataM Intelligence 4Market Research LLP delivers real-time competitive intelligence across autoimmune, immunologic, and rare disease spaces. Our insights span clinical pipelines, regulatory benchmarks, and commercialization strategies for stakeholders in global life sciences.
🔗 Visit: www.datamintelligence.com

